
Summary
Recombineering is a highly efficient and precise method for genetically engineering DNA in vivo. With recombineering one can make gene replacements, deletions, insertions, inversions, and single and multiple point mutations. Gene cloning and gene/protein tagging is also possible. Recombineering is catalyzed by the bacteriophage Red or similar phage homologous recombination systems and can utilize both double- and single-stranded linear DNAs as substrates. All genetic alterations are precise to the base pair as designed by the user and are mediated efficiently by DNA containing target homologies of ~50 bases. These homologies are short enough to be incorporated into commercially available single-stranded DNA oligonucleotides. Since recombineering is directed by sequence homology, not convenient restriction sites, it can be used to create genetic alterations on large DNA molecules such as BACs or chromosomes more precisely than was possible with classical in vitro genetic engineering techniques. Because of this, recombineering has expanded the analytical genetic capability of many organisms. Recombineering with ssDNA can be >100-fold more efficient than with dsDNA as a substrate. Using ssDNA it is possible to achieve recombination frequencies of over 50% of the cells that survive electroporation, thus a selection is not necessary for finding the desired mutations. Key to these high frequencies is avoiding the methyl-directed mismatch repair system, which normally removes greater than 99% of the incorporated changes. Avoiding mismatch repair can be easily achieved, even in wild type cells, with careful oligonucleotide design.
What is recombineering?
The development of genetic engineering revolutionized modern biology, allowing direct manipulation of an organisms DNA. Key to this technology was the discovery of restriction enzymes, proteins that when purified, cut DNA at precise short sequences in vitro. Different DNA molecules, cut with the same restriction enzyme(s), could then be joined together by DNA ligase to generate a recombinant molecule. Such genetically engineered plasmid DNA was routinely transformed into Escherichia coli for further analysis and manipulation. Although these genetic engineering techniques are powerful and continue to be used today, a new highly efficient method for directly modifying DNA within E. coli and other bacteria has been developed in the last ~15 years: recombineering.
Recombineering is in vivo homologous recombination-mediated genetic engineering. The homologous recombination is mediated by bacteriophage-based recombination systems such as the λ Red system, RecET from the Rac prophage, or others. In contrast to classical in vitro genetic engineering, recombineering does not rely on restriction enzymes. Thus, the location of restriction sites is no longer an issue, and the user defines the construct and its location to the base pair. Like genetic engineering, recombineering can be used to make deletions, point mutations, duplications, inversions, fusions and tags. Briefly, recombineering is performed by introducing linear DNA substrates containing the desired change and short flanking homologies to the target DNA into cells expressing the phage-encoded recombination enzymes. These enzymes recombine the linear DNA at the target, yielding recombinant molecules.
Requirements for recombineering.
Recombineering requires that the linear DNA contain homologies to the target DNA of ~50 bases, which are short enough to be built into readily available commercial DNA oligonucleotides (oligos). The linear DNA can be either double-stranded (dsDNA) or single-stranded (ssDNA), and is introduced, generally via electroporation, into the recombineering-competent bacterial cell. For simplicity, we concentrate on the λRed system which consists of three proteins, Gam, Exo and Beta. The Gam protein inhibits the E. coli RecBCD and SbcCD exonucleases that normally degrade all linear dsDNA. Gam is not absolutely required for recombineering but increases the frequency of dsDNA recombination about 10-fold. Exo is a 5’λ3’ double-strand DNA specific exonuclease and is only required for dsDNA recombination. The Beta protein, a ssDNA annealing protein, is the central player in recombineering and is required for both dsDNA and ssDNA-mediated recombination. Importantly, host recombination functions, including the key recombination protein RecA, are not required for recombineering.
In most cases, the target DNA must be replicating: active DNA replication of the target molecule is required for most recombineering reactions. The target DNA can be the bacterial chromosome, plasmids, bacterial artificial chromosomes (BAC) or phage. Thus any DNA, even DNA from human cells cloned into a BAC, can be placed into a recombineering-competent cell and efficiently modified.
Discovery of recombineering
In a seminal paper published in 1998, Kenan Murphy deleted the bacterial recBCD genes, replaced them with the λ Red system, and used linear dsDNA to make gene replacements. These experiments used long, ~1kb, homologies but soon thereafter, Francis Stewart and colleagues demonstrated, using the RecET system, that only 40-60 base pair homologies are required for this type of recombination. We stumbled onto recombineering while doing totally unrelated experiments in the late 1990’s. Our efforts have been to understand and optimize the λ Red system in E. coli and demonstrate the power of oligo recombination.
Recombineering with dsDNA.
Routinely, dsDNA cassettes encoding a drug-resistance are used to make gene knockouts. Typically, the drug-resistant cassette is made by PCR using bi-partite primers. These primers consist of (from 5’λ3’) 50 bases of homology to the target region, where the cassette is to be inserted, followed by ~20 bases to prime the drug-resistant cassette (Figure 1).
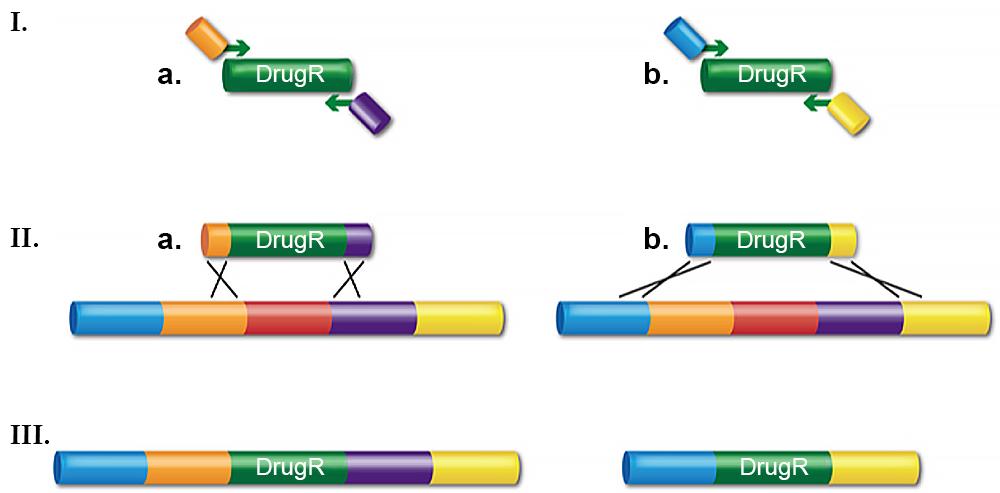
The junction sequence created by the primer design determines precisely how the recombination occurs. For example, the cassette can be inserted between two adjacent bases, the coding sequence of a gene can be replaced to create an in-frame, non-polar knockout, or a whole operon can be deleted. The deletions can range in size from a base pair to >10kb. The PCR fragment is then transformed into electro-competent cells that have been induced for the λ Red system. Having the recombination system under tight regulation yet highly expressed when desired is key for achieving optimal recombination frequencies and preventing unwanted rearrangements. The Red system under control of its own regulated promoter, pL, is much better for this than other systems where the recombination functions are expressed by less tightly controlled promoters.
dsDNA recombineering is very useful for making replacement knockouts and deletions, which are important for many genetic analyses. These recombinants can be found at a frequency of greater than 104 per 108 viable cells ( >104/108 viable). With modifications to the protocol, deletions and point mutations can be made such that there is no drug marker or other alteration left behind. This is accomplished by completing two rounds of dsDNA recombineering using a selection/counter-selection dual cassette. In the first step, the dual cassette is inserted and the (drug) marker is selected. In the second recombination event, the entire cassette is removed by counter-selection. The end result can be a clean deletion, a point mutation created in a gene of interest with no scar left behind, or a tag such as GFP or His. If mutagenic PCR is used to create the linear substrate for the second step, random mutations can be made in the region of interest. The second step can also be done with ssDNA (see below).
in vivo cloning
Recombineering with dsDNA can be used for in vivo cloning by gap repair (Figure 2). Again, a linear substrate is made by PCR but now the DNA fragment contains a plasmid replication origin. The ends of the fragment have homology flanking the region of DNA to be cloned, be it a gene from the chromosome or several introns of a human gene contained on a BAC.
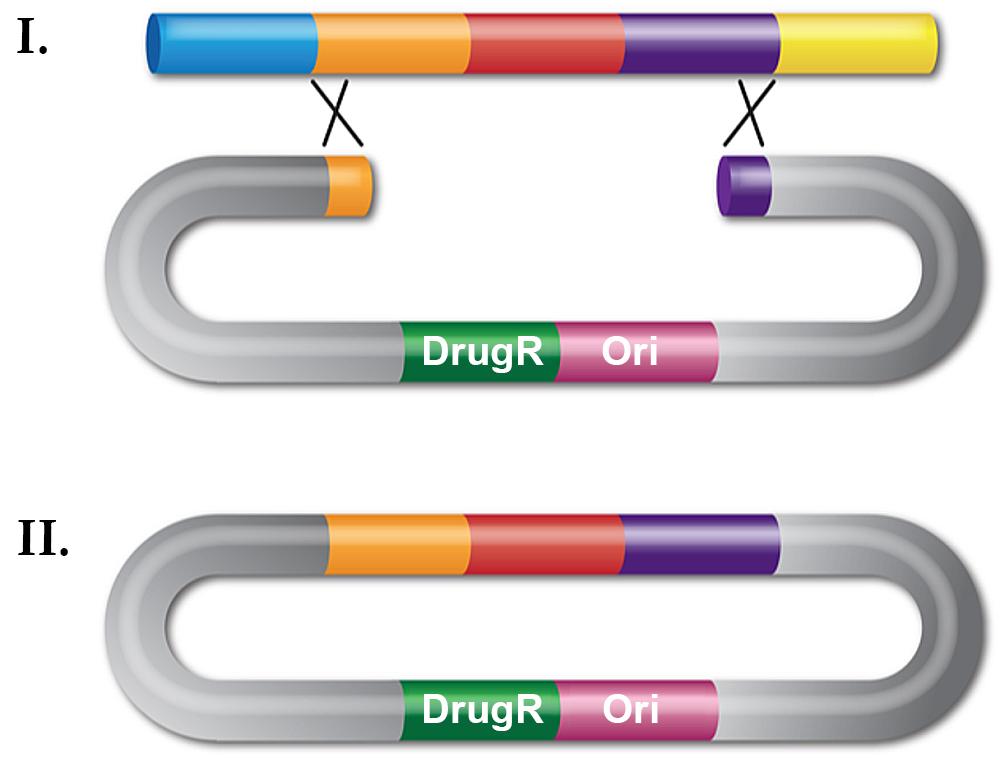
The linear plasmid fragment is introduced by transformation into a recombineering-proficient cell and drug resistance is selected. Plasmid DNA is then isolated and clones screened for the correct insert. In contrast with classical genetic engineering, cloning by retrieval avoids mutations in the cloned gene that arise during PCR, making this technique useful for cloning mutant genes for sequencing. Although this type of recombineering is less efficient than gene replacement, with a frequency around 102/108 viable cells for the λ Red system, it is a very useful cloning technique.
Important note: In 2012, Fu et al. demonstrated that RecET from the Rac prophage is a more efficient system for performing this recombination reaction. In vivo cloing with the RecET system requires the full length RecE protein rather than an N-terminal truncation.
Recombineering with ssDNA
In 2001, Hillary Ellis and colleagues achieved a breakthrough by demonstrating that ssDNA can be used as a recombineering substrate. With a commercial 40-70 base long oligo, precise mutations can be made at a high frequency, typically at 105/108 viable cells, but with some as high as a few percent of the viable cells. Of the three λ Red functions, only Beta is required for recombination with ssDNA. Insight was gained on the mechanism of ssDNA recombination (see below) when it was discovered that of the two complementary oligos that can be used to modify any sequence, the one that corresponds in sequence to the lagging, discontinuously replicated strand is always more efficient, usually by approximately 20-fold.
Optimizing recombineering with ssDNA
With ssDNA recombination it is possible to create single or multiple clustered point mutations (which can create a functional knockout of a gene), small or large (up to 10kb) deletions, and small (10-20 base) insertions such as sequence tags. Multiple genes can be altered by using multiple oligos in the same reaction. Using optimized conditions, point mutations can be made with such high frequencies that they can be found without selection. A crucial discovery came in 2003 when Nina Costantino and Don Court found that avoidance or inactivation of the methyl-directed mismatch repair (MMR) system is key to optimization. In the absence of the MMR system, recombinant cells can be found in over 50% of unselected colonies by a PCR-based screen. The drawback of recombineering in cells mutant for the MMR system, however, is that these cells are mutagenic, allowing additional unwanted genetic changes elsewhere in the genome. The MMR system can be bypassed if the oligo, when paired to the target, creates a C/C mismatch either at or within 6 bases of the desired change. Altering 4-5 bases in a row or altering the wobble position of 4-5 adjacent codons in addition to the desired change will also avoid the MMR system and result in high recombination levels. The latter technique is especially powerful as changes in the wobble position do not alter the protein sequence, thus the technique can be used to mutate essential genes.
In summary, to find optimal recombination levels with ssDNA, an oligo should: 1) be from 40-70 bases in length with the desired change(s) near the middle of the oligo, 2) correspond to the lagging-strand of DNA synthesis, and 3) be designed to avoid the MMR system.
On the mechanism of ssDNA recombination
The current model for ssDNA recombination is that upon entering the cell, the recombining oligo is coated with Beta protein, protecting it from degradation. The Beta-coated oligo then finds and anneals to complementary ssDNA.
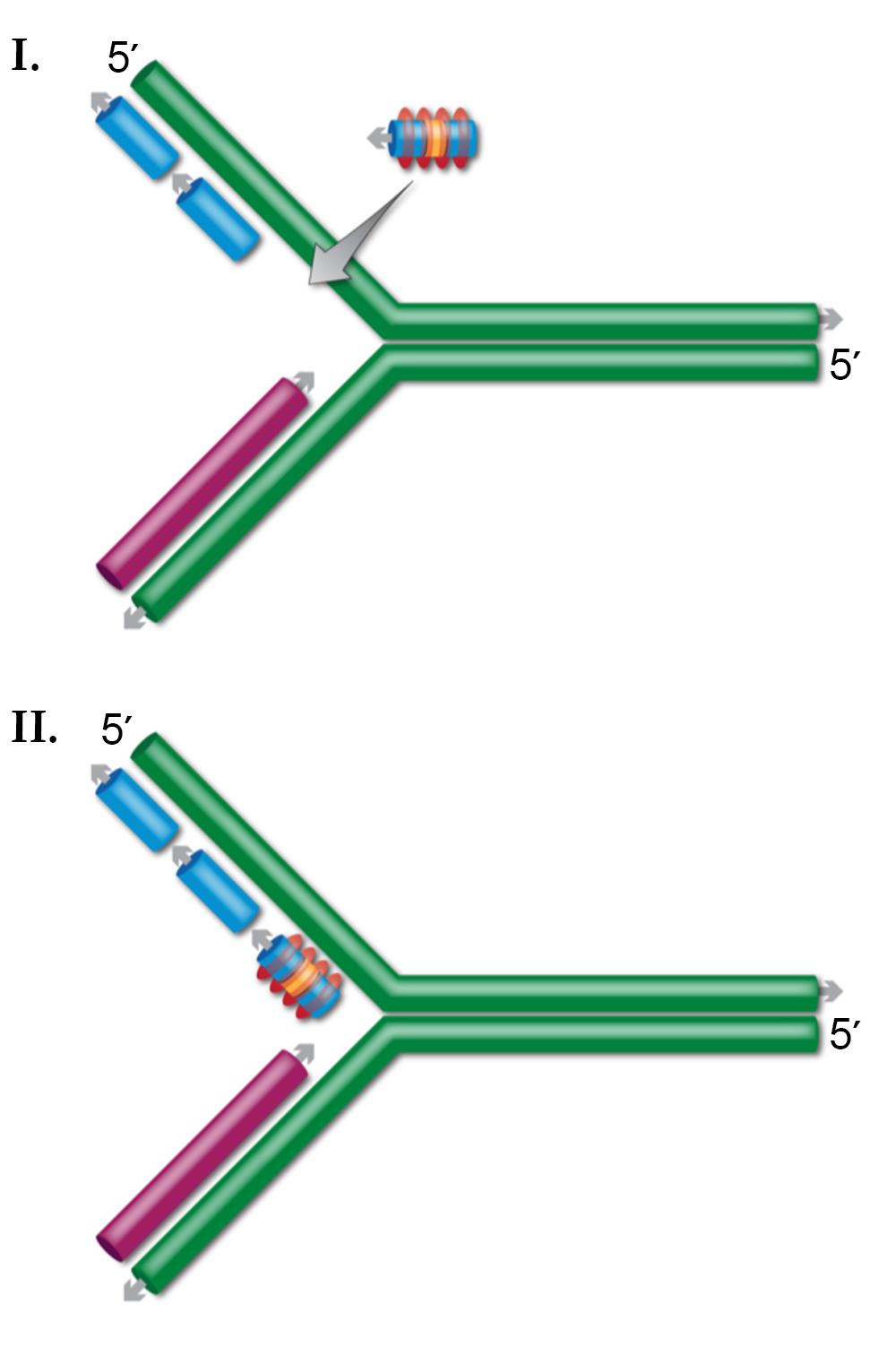
Several lines of evidence indicate that this ssDNA target is present at the DNA replication fork. First, DNA replication is required for ssDNA recombination. Second, of the two complementary oligos that can alter any sequence, the lagging-strand oligo is always more efficient. During DNA synthesis, the lagging-strand has more exposed ssDNA. Third, the MMR system has a large effect on ssDNA recombination, and this system functions at or near the DNA replication fork soon after the fork has passed a given sequence. By this model, after the oligo anneals at the DNA replication fork, DNA ligase joins the oligo to the surrounding Okazaki fragments. As expected, DNA ligase mutations reduce ssDNA recombination.
Moving recombineering to other systems
To date, potential recombineering systems have been identified in over 100 bacteria or bacteriophage that inhabit them (Blast search of Beta and RecT done February 2010), several of which have been cloned and studied in E. coli. The human herpes simplex virus contains analogs as well. These systems hold potential for organisms where the λ Red system does not function. As ssDNA recombination is better understood, less complex, and more efficient than dsDNA recombination, it should be attempted first when developing a new system and the optimized conditions developed in E. coli should be used.
"Recombinase"-independent recombineering
A system for generating ssDNA recombinants has been developed by Bryan Swingle and colleagues. Although not technically “recombineering" as no known bacteriophage functions are involved, similar rules apply. When high concentrations of oligos are transformed into a cell lacking a known recombineering system, ssDNA recombinants are found at a frequency of ~104/108 viable cells. Lagging-strand oligos that avoid the MMR system result in the highest efficiencies. This (known) recombinase-independent ssDNA recombination has so far been demonstrated in P. syringae pv, E. coli, S. enterica, S. flexneri and Y. pseudotuberculosis, and L. pneumophila. It provides another option for creating mutations in organisms where recombineering systems have not yet been developed.