To understand how bacterial and viral infections contribute to human cancers, four NCI at Frederick scientists turned not to the lab bench, but to a computer. The team has created the world’s first—and currently, only—3-D computational approach for studying interactions between pathogen proteins and human proteins based on a molecular adaptation known as interface mimicry.
Invading bacteria and viruses produce proteins that bind with host proteins, causing the activation or inhibition of certain functions in the host’s cells that, in extreme cases, can lead to disorders, chronic diseases, or cancers. Well-known examples include the human papillomaviruses implicated in cervical cancer, as well as Kaposi’s sarcoma–associated herpesvirus, which has been linked to Kaposi’s sarcoma and primary effusion lymphoma.
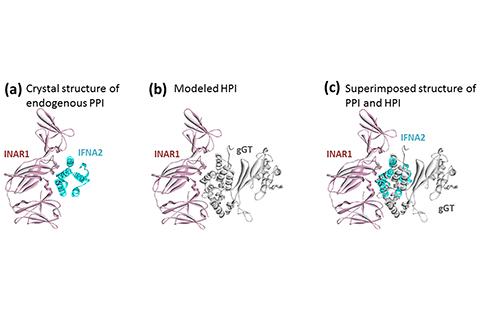
One of the more effective ways that pathogens—especially those associated with cancer—achieve such host–pathogen interactions (HPIs) is through molecular mimicry, in which their proteins “hijack” the appearance of host proteins and use the disguise to avoid the host’s defenses. Interface mimicry, the process by which a pathogen protein copies a host protein’s binding sites, is one type of molecular mimicry.
In their study, the scientists used computer models to examine and predict interface-mimicking HPIs between human proteins and proteins belonging to Helicobacter pylori, a type of gastrointestinal bacteria that has infected over half of the global human population and is associated with gastric cancer.
The researchers believe that their novel approach is highly useful because interface mimicry is one of the most common and efficient forms of pathogenic “hijacking.” The approach can also provide a more detailed perspective than models of larger-scale molecular mimicries, such as structural and sequence mimicry.
“Global structural and sequence similarity is much rarer than interface similarity,” said study author Emine Guven-Maiorov, Ph.D., postdoctoral fellow in the Cancer and Inflammation Program, Center for Cancer Research. “Therefore, interface-based methods would enrich the HPI data more.”
To determine the interface HPIs between H. pylori and human proteins, Guven-Maiorov and her peers studied the 3-D structures of thousands of interface interactions between human proteins, then individually aligned 10 H. pylori proteins with thousands of individual human interfaces to locate interactions that resembled those between the human proteins. Subsequent examinations and validations narrowed the number of matches and eliminated false positives.
From there, the team used the 3-D HPI models to evaluate how H. pylori contributes to gastric cancer. They found that some HPIs block immune cell activation, helping the infection to persist and making it more difficult for the immune system to destroy precancerous cells. Other HPIs increase inflammation, grant H. pylori–infected cells resistance to apoptosis, and increase the infected cells’ capacity to spread to other locations.
The data also allowed them to examine how the HPIs disrupt or shut down the normal protein–protein interactions (PPIs) in the host, which can contribute to cancer by removing the natural functions required to keep cells healthy and/or dispose of unhealthy ones.
In total, the information from the 3-D HPI and PPI models can be used to inform further studies and experimental therapies against H. pylori–associated cancer.
“If we know exactly which host proteins and pathways the pathogen attacks, we can develop small-molecule inhibitors against the interfaces to prevent HPIs,” said Guven-Maiorov. “However, it should be kept in mind that the same inhibitor may abolish both the endogenous host PPI and the exogenous HPI, since their interfaces are very similar.”
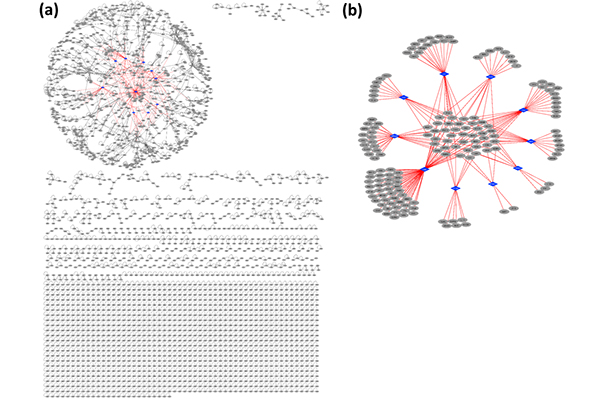
The combined information is also useful for examining HPIs and PPIs on a larger scale. After they had assessed the models, the team integrated all of the 3-D interaction structures into a structural superorganism network—a “web” of all possible HPIs and PPIs that can be used to visualize the effects of all HPIs at once, giving a complete picture of how each interaction affects others.
“Constructing the structural network allows us to determine whether the pathogen targets the hub proteins—proteins that interact with many other proteins—in the network,” said Guven-Maiorov.
She and her fellow scientists learned that multiple H. pylori proteins target hub proteins, making it harder to treat H. pylori–associated cancer by targeting only certain HPIs. However, according to Guven-Maiorov, future studies may help to overcome that hurdle.
“As more HPIs are identified with either experimental or computational techniques, we will have a better understanding of the infectious strategies of pathogens, which will allow [the development of] more efficient therapeutics,” she said.
While that is a promising direction for gastric cancer research, Guven-Maiorov and her collaborators believe that their 3-D computational approach shows promise for more than just H. pylori studies. Results from investigations of other microbial species have proven their approach’s usefulness in modeling other HPIs, and the team is currently planning more studies that will use the approach.
“Our approach can be applied to any pathogenic and commensal proteins, from bacterial, viral, or yeast species,” Guven-Maiorov said.