

Severe forms of neurofibromatosis linked to disruptions in a protein partnership
New research by Frederick National Laboratory (FNL) researchers and their colleagues takes an important step toward explaining complex mutations in a gene called NF1 that is known to cause a disorder known as neurofibromatosis type 1. The study also has potential implications in research against several cancers.
In research described recently in the Proceedings for the National Academy of Sciences, the team focused on one small section of NF1’s protein, neurofibromin, where a handful of these overly potent mutations occur.
Although nominally one disorder, neurofibromatosis can take many, often unpredictable forms, with anything from mild to disfiguring and debilitating symptoms. Likewise, researchers have identified a diffuse set of more than 2,800 mutations in NF1 that can produce it. These errors include small tweaks to the genetic code that have outsized consequences, bringing on severe manifestations of neurofibromatosis.
“One of these mutations can cause patients’ neurofibromin levels to be severely reduced,” says study researcher Jana Ognjenovic, a structural biologist and head of the FNL Advanced Cryo-Electron Microscopy Technology Group. “Our data suggests this drop occurs because the altered versions of the protein ‘poison’ normal protein, leading the cell to discard them both.”
Locked together forever
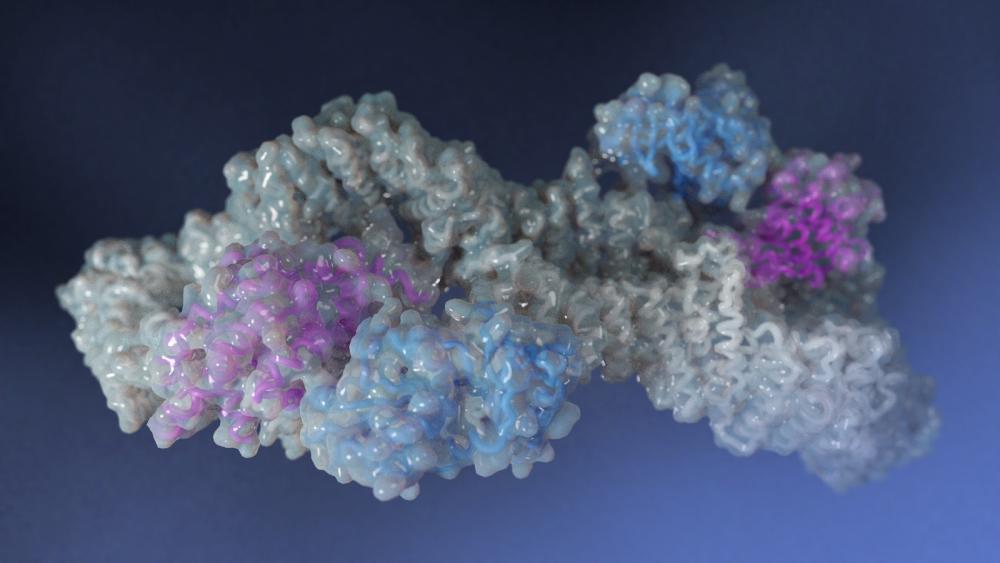
When working as it should, neurofibromin suppresses the formation of tumors, including in neurons and the specialized cells that cover them. Mistakes in this protein can produce skin discoloration and benign tumors around nerve fibers, which appear as bumps on the skin. In more severe cases, patients may have larger, more invasive nerve tumors, as well as bone abnormalities, developmental problems and, occasionally, cancer. These complications often emerge as a child develops. 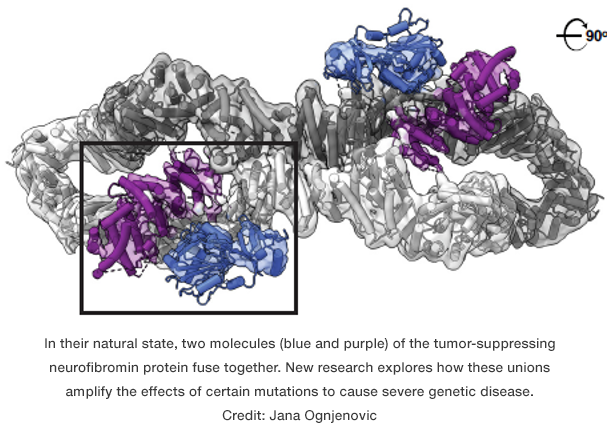
Scientists don't understand why some mutations, but not others, translate to severe forms of the disease.
“One of the problems with neurofibromin is it’s a giant protein, and there are mutations literally all throughout the entire thing that cause disease that can look very different from person to person,” says study author Dominic Esposito, a protein biochemist and head of the Protein Expression Laboratory at FNL. “It's never been clear exactly what's going on.”
In particular, researchers are puzzled by the effects of some small mutations, which alter a single letter of the NF1’s code. If these errors occur in only one of the two copies of the gene that a person carries, they would be expected to render half of the protein useless — a change known to produce, at most, mild symptoms.
But instead, these little errors cut neurofibromin levels more dramatically, causing severe forms of the condition.
Earlier work by Esposito and his colleagues offered a potential explanation: Individual neurofibromin molecules don’t exist on their own, instead they partner up. Together, two molecules bond to form a shape that, from the right angle, calls to mind an infinity symbol.
To explore how the mutations might affect these unions in more detail, the team, including researchers from study author Frank McCormick’s lab at the University of California, San Francisco, focused on a hotspot within neurofibromin where mutations have been linked to spinal nerve tumors and other abnormalities.
A shift in structure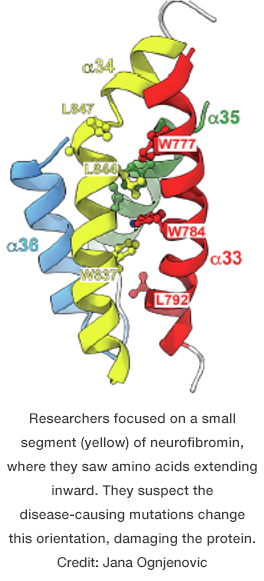
In experiments, they found neurofibromin molecules bearing mutations in this section could still attach to regular versions of the protein. Yet, these half-normal, half-mutant unions proved unstable, and cells treated them as rubbish and degraded them.
To better understand what was happening, Ognjenovic’s group created a detailed, three-dimensional structure of neurofibromin using an advanced imaging technique known as cryo-electron microscopy. This molecular map included the mutation hotspot, itself composed of five building blocks known as amino acids. Within the structure, they saw that these components extended inward, tucked away from the protein’s watery environment.
They realized that the mutations altered the identity of these amino acids, making them compatible with water. Unable to stay where they belong, the mutant amino acids would protrude. This change in shape likely causes the cell to dispose of the affected neurofibromin duos, the researchers hypothesize.
In follow-up experiments, they confirmed that such errors had a devastating effect on neurofibromin. Meanwhile, other changes that did not change the amino acids’ natural aversion to water had little effect on the protein’s viability.
The cancer connection and beyond
While neurofibromatosis type 1 is, on its own, a potentially devastating condition, researchers have an additional motivation for studying neurofibromin: This protein controls the activity of genes, known as RAS, which when mutated drive cancer in the lungs, pancreas, and other organs. Researchers hope that by learning more about neurofibromin, they may find ways to interfere with cancer-promoting changes to RAS genes.
For patients with neurofibromatosis, the study could contribute to improvements in diagnosis, and eventually, perhaps, in treatment. Current therapies are quite limited, but new discoveries about the neurofibromin may point toward new approaches, according to Esposito.
Most significantly, this research establishes a clear link between a set of mutations and certain manifestations of the condition, he says. “That to me opens up the possibility we could make these correlations for other mutations. Maybe understanding these connections will become useful for helping to select therapies.”